Multiple jaw radiopacities and skin soft tissue lesions
Dolphine Oda, BDS, MSc
doda@u.washington.edu
Contributed by
Drs. Jerald Pruner and Mark Egbert
Seattle Children’s Hospital, Seattle, Washington
Can you make the correct diagnosis?
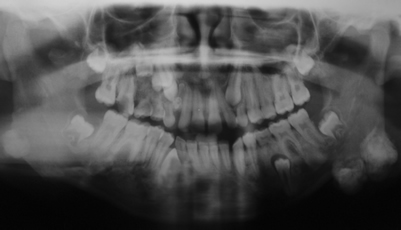
This is a 12-year-old African American girl who presented to the Seattle’s Children’s Hospital with a history of “jaw bumps” that have been present for an unknown period of time.
Sorry! you are incorrect
The etiology of fibrous dysplasia is unknown. The monostotic (one bone or bone complex area) form constitutes approximately 80% of all fibrous dysplasia cases while the polyostotic affects one or more bones with multiple lesions (6). Because of the multiple radiopaque lesions, this case would be considered of the polyostotic type. However, FDs are expansile and disfiguring lesions, whether single or multiple, which differentiate them from the usually flat osteomas of Gardner’s syndrome. Monostotic FD, which involves the jaws, affects males and females equally. It occurs in childhood and at puberty and usually stops growing at age 30. It appears as an asymptomatic swelling of the maxilla or mandible; the maxillary lesion is the most common. It may involve bones other than the maxilla, including the zygoma, sphenoid and others. It is usually unilateral and is known to displace the teeth, but otherwise is firmly seated (6-8). The growth is usually slow, but rapid growth has been described, especially during puberty. The radiographic appearance, especially of the maxilla, is classically described as a ground glass appearance where fine radiopacity is noted. The mandibular lesions are much more deceptive because they tend to vary more, thus making diagnosis with a radiograph difficult. They range from cystic radiolucent forms to classical ground glass radio-opacity (6-7).
Polyostotic fibrous dysplasia constitutes approximately 20% of FD cases (8). Two principle sub-types are described; Jaffe’s and Albright’s syndrome. The latter represents the more severe form with endocrine disturbances (precocious puberty in females). Polyostotic FD is more common in females than males (6-8). Multiple bones are affected including long bones in addition to the jawbones. The clinical presentation depends on the type. Albright’s is associated with hormonal changes, including precocious puberty and cafe au lait spots. Jaffe’s has multiple bones with FD and cafe au lait spots but no hormonal changes. The histology of the bony lesions is similar to that of the monostotic type in which a benign fibro-osseous lesion is identified. The histology of the bony lesions in this case was not supportive of FD. Surgical recontouring of the affected bone may be indicated to address esthetic or functional concerns. This is preferably performed after cessation of growth due to the high incidence of requirement for secondary procedures. Some sources report a tendency for increased growth of the lesion following surgical intervention, but the evidence for this is weak. Even if recontouring is performed in adulthood, multiple procedures are frequently required. This may be due to inadequate removal, continued growth or the ossification of the sub-periosteal hematoma, which is difficult to avoid due to the very vascular nature of the dysplastic bone. Radical excision with primary reconstruction of the affected bone has been suggested, but is not widely accepted. Radiation therapy has been used in the past and is successful in stopping growth of the lesion. Unfortunately, a significant incidence of development of osteosarcoma in the irradiated bone prohibits the use of this modality. Malignancies such as osteosarcoma arising in an area of FD that has not been irradiated have been described, but rarely; these occur mainly in irradiated lesions. The overall prognosis is good with proper clinical management.
Congratulations! You are correct
The combination of multiple osteomas, odontoma and multiple epidermal cysts is highly suggestive of the diagnosis of Gardner’s syndrome, a disease complex condition that affects 1:8000-16,000 births. It presents with multiple osteomas of the jaws, multiple skin epidermal cysts, multiple intestinal polyps and desmoid tumors as the four most common diseases of this syndrome. In addition many other abnormalities are documented, including multiple supernumerary teeth, odontomas, impacted permanent teeth, pigmented eye lesions, and neoplasms (both benign and malignant including sarcomas and neoplasms of the central nervous system, adrenal gland and liver) (1-4). It is autosomal dominant with complete penetrance and variable expressivity (1). The mutated gene is called FAP-GS and is mapped to chromosome 5q21-q22 (1-2). About 25% of the cases are considered to be new mutations. The most important disease in this syndrome is the development of multiple colon and rectum polyps starting at early puberty or before with a mean age of 23-31, while the mean age for cancer formation is 37 (1). By a late age, 100% of the polyps transform to malignancy. By age 30, 50% of patients develop cancer of the polyps and for that reason, it is very important that these patients be closely followed by gastroenterologists for life. Multiple osteomas are also common and about 76% of patients who are FAP positive have osteomas. They affect all bones but are more common in the skull bones including the maxilla, the mandible, and the frontal and ethmoidal sinuses. In one study, up to 93% of the patients had one or more jaw densities. Multiple osteomas of the jaws are present in almost 46% of patients; a few are exophytic, protruding through the mucosa and skin, while most others are in the form of flat calcifications similar to idiopathic osteosclerosis (enostosis) (1-5). They are asymptomatic unless protruding through the jaw as is the case in this patient. They can be associated with torus palatinus and mandibularis. Epidermal cysts are also common in GS patients, and at least half develop them (1-5). The number of these cysts ranges from 1 to as many as 20 with an average of four. Our patient has three at age 12. As in the case of our patient, the epidermal cysts occur on the scalp, face, legs and other areas. The cysts tend to appear around puberty and before the development of the colon polyps. GS patients are also at risk of developing desmoid tumors, especially in the abdominal area following surgery. Desmoid tumors can also occur extra-abdominally and independent of a surgical procedure. Preventive treatment involves removal of the colon to avoid the transformation of the polyps.
References
- Gorlin RJ, Cohen Jr. MM, Hennekam RCM. Gardner’s Syndrome. In: Syndromes of the head and neck, 4th edition. Oxford: Oxford University Press, 2001. p. 437-444.
- Bilkay U Benign osteoma with Gardner syndrome: review of the literature and report of a case. J Craniofac Surg. 2004;15:506-509.
- Daryel EF. Selected pigmented fundus lesions of children. J AAPOS. 2005;9:306-314.
- Capodiferro S, Lacaita MG et al. Multiple and giant mandibular osteomas in a Gardner’s syndrome. Case report. Minerva Stomatol. 2005;54:165-169.
- Nandakumar G, Morgan JA et al. Familial polyposis coli: clinical manifestations, evaluation, management and treatment. Mt Sinai J Med. 2004;71:384-391.
- Parekh SG, Donthineni-Rao R et al. Fibrous Dysplasia. J Am Acad Orthop Surg. 2004;12:305-313.