Fast-growing soft tissue lesion posterior mandible
Can you make the correct diagnosis?
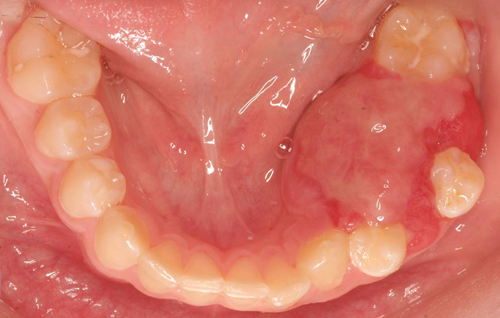
Figure 1 Occlusal view. Photograph taken at first clinical presentation to Children’s Hospital. Note the pink/red, sessile swelling expanding the mandible both buccal and lingual displacing teeth #s 19 and 21. Lingual expansion is more prominent. Tooth # 20 is completely covered by the neoplasm.
Sorry! you are incorrect
Although the age of the patient, the location of posterior mandible, the aggressive behavior and the radiographic findings are suggestive of LCH; the clinical swelling and the histology is not supportive of this condition. The clinical presentation of this case would be consistent with the localized type of LCD. It presents as a solitary osseous lesion anywhere in the body, including the jaw, and especially in the posterior mandible. The most common clinical presentation is that of localized severe periodontitis. More than 50% of cases affect children under 10 years of age, with male predilection. It can be associated with dull pain and tenderness but can also be asymptomatic. Radiographically, it presents as well-demarcated, but not corticated radiolucency with significant bone loss, resulting in the adjacent teeth seemingly “floating in space.” (8) Treatment includes curettage. They have good prognosis.
Sorry! you are incorrect
Although the aggressive clinical presentation and age of the patient are suggestive of Burkitt’s lymphoma, the histology is not supportive of this condition. Burkitt’s Lymphoma, also known as African Jaw Lymphoma, was first described in children of Kampala, Uganda. This lesion is also described worldwide, including in the U.S.A. The African type is a rapidly growing and destructive lesion that occurs as a jaw neoplasm in up to 70% of cases. Its location is usually in the maxilla, but sometimes all four quadrants are involved. The average age of patients with this condition is 7 years. These patients have elevated titer to Epstein Barr Virus (EBV) (9). The non-endemic, non-African type also occurs in children, typically around 10-12 years of age, with male predominance. This type generally involves lymph nodes, lymphoid tissue, and/or visceral organs, especially the abdomen. Jaw involvement is reported but uncommon. These patients occasionally demonstrate EBV antigen titers. Chemothe and radiotherapy are effective treatment modalities.
Sorry! you are incorrect
The clinical presentation and age of this patient can be consistent with fibrosarcoma; however, the histology is not supportive of it. Fibrosarcoma is a rare malignant neoplasm of fibrous connective tissue origin; only 10% of cases occur in the head and neck area. It generally occurs in patients between 20 and 40 years of age, but can occur in infants (10). It is slightly more common in males. It has been reported to occur in the buccal mucosa, maxillary sinus, palate, lips, or the periosteum of the mandible and maxilla. Sarcomas in general present as fleshly, polypoid, rapidly growing, ulcerative swellings that cause facial asymmetry (10) and bone destruction. Treatment depends on the stage of the disease and ranges from surgery to combined surgery and chemotherapy.
Sorry! you are incorrect
The clinical behavior and the age are suggestive of rhabdomyosarcoma, but the histology is not supportive of it. Rhabdomyosarcoma is the most common sarcoma in children. It is a malignant neoplasm of skeletal muscle origin. Three types are described: embryonal. alveolar and pleomorphic. The embryonal and alveolar types are most common in children, while the pleomorphic is most common in adults. All types of rhabdomyosarcomas present as exophytic, polypoid, ulcerated, and rapidly growing lesions (11). This condition may occur at birth, in children, in teenagers or in young adults; it is rare after the age of 45 (11). The head and neck area is a common location for this neoplasm which constitute 40% of all cases. The orbit is the most common location, followed by the nasal cavity, the oropharynx and the oral cavity (11). Within the oral cavity, the palate is the most common location. Treatment depends on the stage of the disease and ranges from surgery to combined surgery and chemotherapy.
Congratulations! You are correct
Aggressive fibromatosis represents a group of fibrous neoplasms with various clinical behaviors and histologic presentations. Aggressive fibromatosis is known by many names, including juvenile aggressive fibromatosis, infantile desmoid fibromatosis (1), and nonmetastasizing fibrosarcoma (2). The last nomenclature best describes the behavior of this lesion; it is a benign, but locally infiltrative and aggressive neoplasm with a potential for multiple local recurrences, but no potential for metastasis. Aggressive fibromatosis can be sporadic or familial such as those associated with Gardner’s syndrome which tends to occur more in the abdominal area, and rarely in the head and neck area (1, 3). Some investigators consider aggressive fibromatosis in children to be a separate entity than that of adult fibromatosis (1, 5). Head-and-neck aggressive fibromatosis usually occurs in children under 20 years of age (1). It is more prevalent in adult females by a ratio of 2:1 and in children more in males by 2:1 (5, 7). It typically presents as a firm, painless, rapidly growing asymptomatic mass. Occasionally, it is slow-growing, but it is destructive in either case. It can destroy bone, infiltrate adjacent structures, displace teeth, and induce periosteal reaction, rendering it clinically and radiographically difficult to differentiate from a sarcoma. It presents in multiple locations within the oral cavity, including the gingiva, lips and other oral soft tissue areas. Researchers have proposed several theories regarding the etiology of aggressive fibromatosis, which is currently unknown. Hormonal, viral, chromosomal, and, occasionally, trauma-induced etiologies have all been described. Trauma-induced etiology is often described in the area of caesarean-section scars (4). There is a legitimate body of evidence to suggest that estrogen promotes the growth of fibromatosis, particularly in pregnant females (4). Association with the FAP (Gardner’s Syndrome) gene is also well documented (5). The clinical behavior of aggressive fibromatosis is unpredictable and ranges from frequently recurring to spontaneously regressing and for that reason, a variety of treatment modalities have been employed. Surgery, which is the treatment of choice, ranges from complete removal with clean margins to debulking of the lesion to avoid loss of vital structures. The latter method is often necessary for oral cavity lesions. In the present case, care was taken during surgery to avoid sacrificing the lingual nerve; therefore, the surgeons opted for the more conservative approach of debulking of the primary lesion and very close watch of the surgical area. Several non-surgical alternative treatments have also emerged, such as radiotherapy, chemotherapy, hormonal and biological manipulation, and non-steroidal anti-inflammatory agents. Surgery combined with radiotherapy or chemotherapy has been used in recurring and persistent lesions. Radiotherapy is a secondary treatment method to surgery, and is sometimes used alone (6). However, concern over its possible adverse long-term effects, especially in children, makes it a less desirable mode of therapy. Chemotherapy, with single or multiple agents, and neoadjuvant or maintenance therapy have also been used (6). In children, Chemotherapy is associated with long-term adverse effects and occasional serious complications and even death (6). Some cases have been successfully treated with alternative treatments such as estrogen antagonists and non-steroidal anti-inflammatory drugs (5). Prognosis and recurrences are difficult to predict. There is a range of 20%-70% recurrence rate for aggressive fibromatosis in children, mostly recurring within the first five years (7). Some studies suggest that positive margins may be the only predictor for recurrence, if any (6). Other adverse prognostic factors may include age of less than 18 years, recurrent disease, and surgical treatment alone (5-7).
References
- Coffin CM, Dehner LP. Pediatric Soft Tissue Tumors. 1997; 133-178.
- Donahue WB, Malexos D, Pham H. Aggressive fibromatosis of the maxilla. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1990; 69:420-426.
- Jones IT, Jagelman DJ, et al. Desmoid tumors in familial polyposis coli. Ann Surg. 1986; 170:109-121.
- Hayry P, Reitamo JJ, et al. The desmoid tumor II. Analysis of factors possibly contributing to the etiology and growth behavior. Am J Clin Pathol. 1982; 77:674-680.
- Fletcher CDM. Diagnostic Histopathology of Tumors, Volume 2. 2000; 1498-1500.
- Chalmers AJ, Gillham CM et al. Nuchal aggressive fibromatosis in childhood: two instructive case reports. Clin Oncol. 2001; 13:378-383.
- Roychoudhury A, Parkash H et al. Infantile desmoid fibromatosis of the submandibular region. J Oral Maxillofac Surg. 2002; 1198-1202.
- Bonet J, Manuel MJ et al. Eosinophilic granuloma of the jaws: a report of three cases. Med. Oral. 2001; 6: 218-224
- Cakmak SS, Soker M et al. Non-African Burkitt’s lymphoma manifesting at the jaw and as a right orbital mass in a child. J Pediatr Ophthalmol Strabismus. 2003; 40: 306-308.
- Lo Muzio L, Mignogna MD et al. A rare case of fibrosarcoma of the jaws in a 4-year-old male. Oral Oncol. 1998; 34: 383-386
- Chigurupati, R, Aflatooni, oda, D and Myall, R. Rhabdomyosarcoma of the head and neck in children, review of literature and addition of four cases. J Oral Oncology. 2002; 38: 508-515.