Multiple Intraoral Soft Tissue Swellings
Can you make the correct diagnosis?
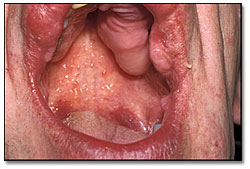
This patient presented with dental pain and multiple soft tissue swellings in the mouth, head and neck area, as well as in other parts of the body.
Sorry! you are incorrect
Tuberous Sclerosis Complex is a common neurocutaneous syndrome with multiple skin nodules. It is autosomal dominant with 70% spontaneous mutation. TSC is characterized by seizures, multiple cutaneous hamartomatous growths and central nervous system disorders such as mental retardation. The multiple cutaneous growths can involve organs other than the skin. Two types of this syndrome are described and the genes have been mapped to chromosome 9 for TSC1 and chromosome 16 for TSC2. This syndrome presents with a spectrum of presentations ranging from mild and undiagnosed to severe cases. The prevalence varies from 1:23,000 to 1:170,000; autopsy studies indicate its incidence rate in the general population to be as high as 2.5%.
Among the clinical manifestations are skin lesions such as angiofibromas, mostly occurring around the nasolabial fold. They are smooth-surfaced red nodules; they can also involve the nails. Hypopigmentation (ashleaf spots) also occurs and sometimes requires special light for identification. The latter is the first symptom to appear—sometimes at birth—and is the most common presentation. Other significant lesions include subependymal nodules of the brain; 90% of patients with this condition develop seizures and 60% are mentally deficient. Cardiac rhabdomyomas are described in 30% of patients. Renal, ocular and skeletal abnormalities are also described. The histology and the clinical presentation of this case are not supportive of TSC (1).
Sorry! you are incorrect
This is a rare disease complex involving the skin, breast, gastrointestinal tract and thyroid. It is autosomal dominant and the gene has been mapped to chromosome 10q23. Skin lesions are papillomatous and smooth surfaced nodules and occur in about 99% of patients with this condition. They occur mostly on the face, particularly around the eyelids, nose, mouth and ears. They may also affect the arms and hands. The nodules are mostly trichilemmomas, of hair follicle origin. Breast diseases include fibrocystic disease and some malignant neoplasms. Thyroid lesions include goiter, follicular adenocarcinoma, and polyps; these are typically benign in behavior. The histology and the clinical presentation of this case are not supportive of TSC (1, 2).
Congratulations! You are correct
Neurofibromatosis type I (NFI) was first described by Frederick von Recklinghausen in 1882 and for that reason has also been known as von Recklinghausen’s Neurofibromatosis (1). It is a common autosomal dominant disease affecting 1:2,500 to 1:3,000 newborn with 50% mutation and almost 100% penetrance (1). The genetic mutation of NFI has been mapped to chromosome 17q11.2. Nine types have been described; types I and II are well delineated while the other types are rare and not well studied (1). Type I accounts for more than 90% of cases, followed by type II, which is also known as acoustic type, where bilateral acoustic neuromas are described leading to hearing loss starting as early as the teenage years. Type II is much less common and affects 1 in 40,000 individuals (1, 4). The genetic mutation of NFII is mapped to chromosome 22 (4).
The main diagnostic features of neurofibromatosis type 1 are cutaneous neurofibromas, café-au-lait spots (six or more of 1.5 cm size and larger), axillary freckling (Crowe’s sign), Lisch nodules, and several other lesions. The latter include lesions involving the central nervous system such as mental retardation in about 8% of patients, the cardiovascular system such as pulmonic valvular stenosis, the skeletal system such as scoliosis and the endocrine system such as sexual precocity. Eyes are also involved such as neurofibromas of the eyelids and Lisch nodules of the iris. The cutaneous neurofibromas occur at birth and are present at puberty in 60% of cases. They increase in number with time and can number from a few to in the thousands. They can be present on the skin or in internal organs such as the heart, brain or the gastrointestinal system. They can be small and demarcated or large and pendulous (1).
Oral soft tissue manifestations of neurofibromatosis can occur in 8-66% of cases, and jaw lesions in 58% of patients (5). The most common lesion is neurofibroma, which occurs mostly in the soft tissue, especially the tongue. It may also affect the jaw bones. Widening of the mandibular foramen and inferior alveolar canal, as is the case in our patient, are also described. Other lesions include fungiform papillae, hyperplasia of the soft palate and displaced and unerupted teeth, also features seen in our patient.
Histologically, the neurofibromas can be the conventional, more common type of haphazardly arranged neural tissue in a relatively demarcated nodule or can be the plexiform type where nerve tissue is arranged in lobules. The latter histology is pathognomonic of neurofibromatosis. In the head and neck area plexiform neurofibroma can be associated with the trigeminal nerve branches.
Treatment in Neurofibromatosis patients is primarily for esthetic and functional reasons. Scalpel surgery, laser or dermabrasion treatments have been used for the removal of skin neurofibromas. Malignant transformation has been described in about 8% of patients, most of which are peripheral nerve type malignancies which tend to be aggressive. Other malignancies have been described including leukemia, rhabdomyosarcoma, Wilm’s tumor, Pheochromocytoma and several others (1). The general dentist may be the first person to make the primary diagnosis of Neurofibromatosis as was the case with our patient. Therefore, it is important to know of this disease and its characteristic clinical manifestations. These patients should be referred to physicians for further clinical workup, including genetic workup.
Treatment
Tooth #30 was extracted. The patient chose to save tooth #31 with endodontic treatment and restoration. One of the buccal nodules was biopsied.
References
- Gorlin R, Cohen MM et al. Syndromes of the Head and Neck. 1990 Oxford University Press.
- Tsou HC, Ping XL et al. The genetic basis of Cowden’s syndrome: three novel mutations PTEN/MMAC1/TEP1. Human Genet. 1998; 102: 467-473
- Lips CJM, Hoppener JWM et al. Medullary thyroid carcinoma: role of genetic testing and calcitonin measurement. Annals clinical Biochem. 2001; 38: 168-179.
- Twist EC, Ruttledge MH et al. The Neurofibromatosis type 2 gene is inactivated in schwannomas. Human Mol. Genetics. 1994; 3: 147-151.
- D’Ambrosio JA, Langlais RP et al. Jaw and skull changes in Neurofibromatosis. 1988 Triple O (Oral Surg); 66: 391-396.
Sorry! you are incorrect
These are rare disease groups affecting the endocrine system. Three types have been described. Some are inherited as autosomal dominant while others develop as a result of mutation. MEN syndrome type 2b is the most significant type to dental practitioners. The gene for this type has been mapped to chromosome 20p12.2. Lesions appear as early as infancy, causing problems with feeding and normal thriving (1). During the first decade, multiple small mucosal nodules occur in the oral cavity on the anterior tongue, lower lip, and bilateral corner of mouth. These nodules are highly characteristic of the disease. They may also occur on the eyelids and conjuctiva, and represent multiple neuromas which are histologically made up of hyperplastic peripheral nerve fibers. Multiple melanotic skin lesions have been described in these patients. Patients have marfanoid features, a thick lower lip, and an everted upper eyelid. They also develop pheochromocytoma, profuse sweating, diarrhea, and severe hypertension. In addition, they often develop medullary carcinoma of the thyroid at around 18-25 years of age, but this aggressive neoplasm has been described in patients as young as 23 months. MEN 2b patients demonstrate high levels of catecholamines and calcitonin if pheochromocytoma and medullary carcinoma are present. Preventive removal of the thyroid gland is recommended (1, 3). The clinical presentation and histology of this case are not supportive of MEN 2b syndrome.