Symmetrical Bilateral Mandibular Multilocular Radiolucencies
Can you make the correct diagnosis?
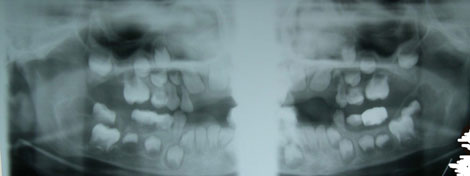
This is a 5-year-old male who was referred to Children’s Hospital and Regional Medical Center Oral and Maxillofacial Surgery Department by Dr. GB Givler of Helena, Montana.
Sorry! you are incorrect
Central giant cell granuloma (CGCG) is described as a non-neoplastic process and yet can behave in a very aggressive and expansile manner, destroying bone and displacing teeth. Over 60% of CGCG cases occur in patients younger than 30 years of age, with twice as many occurrences in females. CGCG is classified into aggressive and non-aggressive types. The aggressive type tends to occur in younger patients with disfiguring results, especially after surgery. Over 70% of cases occur in the mandible anterior to the first molar tooth. This lesion has also been described in other cranio-facial and small long bones such as those of the hands and feet (1-3). The usual treatment for CGCG is surgery, ranging from curettage and en bloc to resection. The latter is used in aggressive and recurring cases. However, treatments alternative to surgery are also present and include: steroid injections (1,3), the most successful alternative treatment thus far, which require injections weekly or every 2-3 weeks, have no known side effects (even in children), and are the least expensive alternative treatment; calcitonin injections or nasal spray (2), which require daily injections or a nasal spray of salmon calcitonin for about a year and are safe for pregnant females; and interferon alfa-2a injections (2), which are administered 2-3 times per week for several months and are the most expensive alternative treatment. Although the histology and some of the clinical and radiographic findings are consistent with CGCG, the fact that this condition is symmetrically bilateral is not supportive of it. Also, CGCG tends to occur anterior to the first permanent molar. This lesion was growing posteriorly into the ramus, unusual for CGCG
Sorry! you are incorrect
Three types of hyperparathyroidism are described: primary is the most common; secondary and tertiary are the least common. All three types are characterized by excess parathyroid hormone production. Primary hyperparathyroidism is characterized by hypercalcemia and hypophosphatemia, secondary hyperparathyroidism is characterized by hyperphosphatemia and mild hypocalcemia, and tertiary hyperparathyroidism is characterized by hypercalcemia. Since primary hyperparathyroidism is the most common type, it will be described here in more detail than the other two types.
Primary hyperparathyroidism is usually caused by adenoma of the parathyroid gland, but is sometimes a result of hyperplasia (in about 10% of cases) and is rarely caused by adenocarcinoma (4). Secondary hyperparathyroidism is a sequelae to chronic renal disease (renal osteodystrophy). Primary hyperparathyroidism is 3 times more common in females than males and typically occurs in patients in their fifties and older. The clinical presentation is characteristically referred to as bones, stones, groans and moans—affecting multiple organs including bones, kidney stones, gastrointestinal system groans, muscles and the central nervous system moans. Bone lesions are painful multiple unilocular and multilocular radiolucencies that affect the fingers and the skull, including the jaw bones. They are called ‘brown tumors’ because of the deep chocolate brown color of the specimens resulting from hemorrhage and hemosiderin pigmentation. Stones are result of hypercalcemia affecting the kidneys and skin. Groans are related to intestinal ulcers and constipation, and moans are related to alteration in the central nervous system such as depression and sometimes seizures (4). This patient did not have alteration in his calcium/phosphate levels. Histologically, the morphology is suggestive of brown tumor. However, the clinical or radiographic presentation are not consistent with brown tumor of hyperparathyroidism.
Congratulations! You are correct
Cherubism is a benign condition that involves bilateral swelling of the maxilla and/or the mandible that was first described by Jones in 1933 as a familial multilocular cystic disease of the jaws. Most of the cases are hereditary, autosomal dominant with 100% penetrance in males and 50-70% penetrance in females (5). For the inherited type, the gene has been identified as SH3BP2 which is mapped on chromosome 4p16.3. A non-hereditary form is also described (6) but is rare, an example of the present case where non of the siblings and parents have the disease. Both types of cherubism occur during childhood in patients as young as 1 year of age with an average age of 7. They regress after puberty and in rare occasions may continue to grow even after age 20. They characteristically present with symmetrical bilateral cheek swelling involving the angle of the mandible. Mandible is more commonly involved, sparing the condyle and involving the coronoid process (5). However, unilateral involvement with positive family history has been described. It can affect all four quadrant and maxillary sinuses. The fullness of the maxilla and sinuses leads to the upward rotation of the eyes with a cherubic look—hence, the name “cherubism”—due to orbital floor involvement. Diplopia is not present. Radiographically, there is multilocular radiolucency with corticated border. The locules can be well-defined with clear septa formation, as is the case here, and can be blurred with just scalloped borders. The latter is seen during regression of the disease. Cherubism can be expansile and disfiguring. It may thin the bone to a point of perforation, but fracture is rare. Early resorption and exfoliation of the deciduous teeth is described as is the case with this patient. Displacement of permanent teeth has also been described. The histology of cherubism consists of granulation tissue stroma, giant cells and blood vessels with hyaline deposits. The histology of the present case fits the characteristics well. Cherubism can spontaneously regress after puberty. Loss of loculation, of expansion and filling of the bony spaces with normal bone are signs of regression. However, if the lesion is large and disfiguring, surgical intervention is considered knowing that this lesion can spontaneously regress. Cherubism is also known to be associated with syndromes such as Noonan’s syndrome and is described to occasionally affect other bones such as the ribs (5-6).
Treatment
The incisional biopsy was performed under general anesthesia induced and maintained by an anesthesiologist. Approximately 8 cc of 0.05% Marcaine with epinephrine was injected, with bilateral inferior alveolar nerve block and local infiltration. An incision along the right mandibular ramus with extension along the buccal aspect of tooth #T was made. Tooth #T was removed and a biopsy of a one cubic cm of the right mandibular ramus was performed. These tissues were similarly submitted for histopathologic examination. The surgical site was copiously irrigated and a flap was reapproximated and closed with 3-0 Vicryl sutures. Teeth #s K and L were similarly extracted and the wound was copiously irrigated and the tissues were closed with 3-0 Vicryl suture. Gauze packs were placed on the surgical sites for hemostasis. The patient was returned to the care of the anesthesiologist for recovery and was extubated in the operating theater without incident. He was discharged with Tylenol with Codeine elixir 5 cc p.o. q. 4 to 6 hours p.r.n. pain and peridex rinse, 10 cc Swish and Expectorate X30 seconds t.i.d. A three day postoperative follow-up visit demonstrated that the wounds were healing well and that he had an approximate 20-mm mouth opening with mild discomfort and intact neurosensory innervation. A 3-month follow-up visit showed no significant expansion of the bilateral mandibular lesions with some potential shrinkage of the bilateral lesions, especially the right mandibular area where the surgical intervention was greatest. The patient will continue to be monitored. The possibility of chemotherapeutic interventions using alpha interferon was discussed and agreed to be used only if the lesions were to dramatically deteriorate.
References
- Carlos R, Sedano HO. Intralesional corticosteroids as an alternative treatment for central giant cell granuloma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2002; 93(2):161-6.
- O’Regan EM, Gibb DH, Odell EW. Rapid growth of giant cell granuloma in pregnancy treated with calcitonin. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 200; 92(5):532-8.
- Collins A. Experience with anti-angiogenic therapy of giant cell granuloma of the facial bones. Ann R Australas Coll Dent Surg 2000; 15:170-5.
- Yamazaki H, Ota Y et al. Brown tumor of the maxilla and mandible: progressive mandibular brown tumor after removal of parathyroid adenoma. J Oral Maxillofac Surg. 2003; 61:719-722.
- Langlais RP, Langland OE, Nortje CJ. Diagnostic imaging of the jaws. Williams & Wilkins. 1995. Pages 367-370.
- Ozkan Y, Varol A et al. Clinical and radiological evaluation of cherubism: a sporadic case report and review of the literature. Int J Pediatr Otorhinolaryngol. 2003; 67:1005-1012.
- Fardon PA, Norris D.J et al. Analysis of 133 meioses places the genes for nevoid basal cell carcinoma (Gorlin) syndrome and Fanconi anemia group C in a 2.6-cM interval and contributes to the fine map of 9q22.3, Genomics. 1994; 23: 486-489.
- Shear M. Odontogenic keratocysts: natural history and immunohistochemistry. Oral Maxillofacial Surg Clin N Am. 2003; 15: 347-362.
- Stoelinga, PJW. Excision of the overlying, attached mucosa, in conjunction with cyst enucleation and treatment of the bony defect with Carnoy solution. Oral Maxillofacial Surg Clin N Am. 2003; 15: 407-414.
- Oda D, Rivera V et al. Odontogenic keratocyst: the northwestern USA experience. J Contemp Dent Pract. 2000 Feb 15; 1(2): 60-74.
Sorry! you are incorrect
Odontogenic keratocyst (OKC) is an aggressive odontogenic cyst and is known for its rapid growth and its tendency to invade the adjacent tissues, including bone. The majority of OKC occur as single and isolated lesions that are not associated with a syndrome. The profile of these cysts does not fit this case. However, OKC can also be present in multiple as part of bifid-rib basal cell nevus syndrome (Gorlin syndrome). This syndrome is an autosomal dominant disorder arising from defects on chromosome 9q23.1-q31 (7), and is often identified in juvenile kindred under 10 years of age (7). When so associated, the cysts are frequently multiple and the patient is generally young such as in the present case (7-10). They present more frequently in the mandible, can be multilocular and tend to grow in an anterior-posterior manner extending into the ramus (8). As described In case of the month March 2004, OKCs have a tendency to recur and behave destructively. They are known to have a high recurrence rate, ranging from 13% to 60% (7-10). Complete surgical removal is the treatment of choice. Surgery combined with Carnoy’s solution or liquid nitrogen cauterization has been effective in reducing recurrence rate (9-10). At times, adjacent or associated teeth are extracted in the interest of complete removal. Some investigators advocate marsupialization and occasionally resection of the more aggressive cysts that tend to perforate buccal and lingual bone (10). Resection is a rare modality of treatment. Most cysts recur within the first three years while others may recur as late as after 16 years (10). Conservative surgical removal and long-term follow-up is the treatment of choice by most clinicians. They have a specific histology which was not consistent with the histology of this case.