September 2010: Multiple Radiolucencies of the Maxilla and Mandible
Can you make the correct diagnosis?
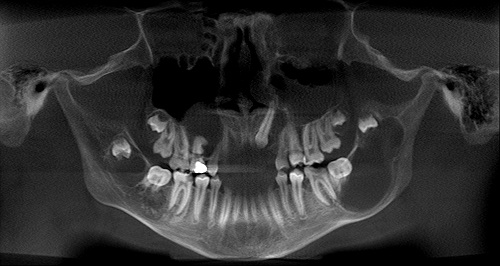
This is a 14-year-old white male who was referred to South Sound Oral Surgery by his general dentist for extraction of a painful, carious mandibular left first molar.
Sorry! you are incorrect
The age of the patient and the multiplicity of the bony lesions lend themselves to a diagnosis of cherubism. However, cherubism starts at a much younger age and ends at puberty (ages 1-14, with an average age of 7). Cherubism tends to feature multilocular and expansile radiolucencies, usually bilateral, in the posterior mandible but not associated with impacted molars. This patient has bilateral posterior mandible radiolucencies that are clearly associated with impacted teeth and are only mildly expansile (or not at all), making this unlikely to be a case of cherubism. The histology is also not supportive of cherubism.
Cherubism is a benign condition that involves bilateral swelling of the maxilla and/or the mandible. It was first described by Jones in 1933 as a familial multilocular cystic disease of the jaws. Most cases are hereditary: autosomal dominant with 100% penetrance in males and 50-70% penetrance in females (1). A non-hereditary form is also described (2). Both types of cherubism occur during childhood in patients as young as 1 year of age with an average age of 7. They regress after puberty and in rare occasions may continue to grow even after age 20. They characteristically present with symmetrical bilateral cheek swellings involving the angle of the mandible. The mandible is more commonly involved, sparing the condyle and involving the coronoid process (1). However, unilateral involvement with a positive family history has been described. It can affect all four quadrant and maxillary sinuses. The fullness of the maxilla and sinuses leads to the upward rotation of the eyes with a cherubic look—hence, the name “cherubism”—due to orbital floor involvement. Diplopia is not present. Radiographically, there is multilocular radiolucency with corticated border. The locules can be well-defined with clear septa formation, and can be blurred with just scalloped borders. The latter is seen during regression of the disease. Cherubism can be expansile and disfiguring. It may thin the bone to a point of perforation, but fracture is rare. Early resorption and exfoliation of the deciduous teeth is described. Displacement of permanent teeth has also been described. The histology of cherubism consists of granulation tissue stroma, giant cells and blood vessels with hyaline deposits. Cherubism can spontaneously regress after puberty. Loss of loculation, of expansion and filling of the bony spaces with normal bone are signs of regression. However, if the lesion is large and disfiguring, surgical intervention is considered, even while knowing that this lesion can spontaneously regress. Cherubism is also known to be associated with syndromes such as Noonan’s syndrome and is described to occasionally affect other bones such as the ribs (1-2).
Sorry! you are incorrect
The multiplicity of the bony lesions suggests that hyperparathyroidism should be included on the differential diagnosis. However, the age and sex of the patient make hyperparathyroidism an unlikely diagnosis. Another reason that would be hyperparathyroidism unlikely is the fact that all of these lesions are associated with impacted teeth and that would speak against hyperparathyroidism. The histology is not supportive of hyperparathyroidism either.
Three types of hyperparathyroidism are described: primary, which is the most common; secondary; and tertiary, which is the least common. All three types are characterized by excess parathyroid hormone production. Primary hyperparathyroidism is characterized by hypercalcemia and hypophosphatemia, secondary hyperparathyroidism is characterized by hyperphosphatemia and mild hypocalcemia, and tertiary hyperparathyroidism is characterized by hypercalcemia. Since primary hyperparathyroidism is the most common type, it will be described here in more detail than the other two types.
Primary hyperparathyroidism is usually due to an adenoma of the parathyroid gland, but is sometimes a result of hyperplasia (in about 10% of cases) and is rarely caused by adenocarcinoma (3). Secondary hyperparathyroidism is a sequel to chronic renal disease (renal osteodystrophy). Primary hyperparathyroidism is 3 times more common in females than males and typically occurs in patients in their fifties and older. The clinical presentation is characteristically referred to as “bones, stones, groans and moans”—affecting multiple organs including the bones, the kidneys (“stones”), the gastrointestinal system (“groans”), and the muscles and central nervous system (“moans”). The associated bone lesions are painful multiple unilocular and multilocular radiolucencies that affect the fingers and the skull, including the jaw bones. They are called ‘brown tumors’ because of the deep chocolate brown color of the specimens resulting from hemorrhage and hemosiderin pigmentation. The kidney stones are the result of hypercalcemia affecting the kidneys and skin. The “groans” are related to intestinal ulcers and constipation, and the “moans” are related to alteration in the central nervous system such as depression and sometimes seizures. This patient did not have alteration in his calcium/phosphate levels. Histologically, the biopsy material was not consistent with brown tumor of hyperparathyroidism (3).
Congratulations! You are correct
The diagnosis of multiple odontogenic keratocysts almost classifies this patient’s case as nevoid basal cell carcinoma syndrome (NBCCS), which is also known as bifid rib basal cell nevus syndrome, basal cell nevus syndrome, Gorlin syndrome, Gorlin-Goltz syndrome, and other names. Nevoid basal cell carcinoma syndrome is an autosomal dominant inheritance affecting one in 50,000 to one in 150,000 individuals in the US. The NBCCS gene is due to a mutation in the Patched (PTCH) gene on chromosome 9q22.3-q31 (4-7). There is evidence to implicate this gene in isolated non-syndrome-associated OKCs, but it is known that one third to one half of cases represent a new mutation. NBCCS has almost one hundred clinical features divided into major and minor categories (4,6); the presence of two major features or one major feature with two minor features would classify the patient’s case as NBCCS (6).
Major criteria include (4, 6):
- More than two basal cell carcinomas (BCCs) or one BCC in patients younger than 20 years of age.
- Multiple histologically confirmed odontogenic keratocysts
- Three or more palmar or plantar pits
- Bilamellar calcification of the falx cerebri
- Bifid, fused, or markedly splayed ribs
- First-degree relative with NBCCS
Minor criteria include (4, 6):
- Macrocephaly, after adjustment for height
- Congenital malformations, such as cleft lip or palate
- Frontal bossing, coarse facies, and moderate or severe hypertelorism
- Other skeletal abnormalities, such as Sprengel deformity, marked pectus deformity, and marked syndactyly of the digits
- Radiologic abnormalities, such as bridging of the sella turcica, vertebral anomalies, modeling defects of the hands and feet, or flame-shaped lucencies of the hands & feet
- Ovarian fibroma or medulloblastoma
As demonstrated above, the clinical features of this syndrome encompass a variety of structures including the jaw bones, skin, central nervous system and the skeletal system. Basal cell carcinomas, Odontogenic keratocysts and palmar and plantar pitting are the three most common manifestation of the syndrome (4-6).
Basal cell carcinomas usually appear between the ages of 13 and 35 but have been described in patients as young as two years of age (4, 6). The number of BCCs varies from just a few to hundreds, and white patients with the syndrome tend to develop more BCCs than black patients with the syndrome (4, 6). The BCCs affect both sun-exposed (face i.e. nose, eyelids, cheeks) and non-sun-exposed skin (i.e. back, chest, arms) and are usually above the waist line.
Over 75% of NBCCS patients present with odontogenic keratocysts, usually in multiples (4-7). About one third present with a single cyst that is followed by others over a twenty-year period. OKCs in general tend to occur more in the mandible than in the maxilla by a ratio of about 3:1. OKCs in patients with NBCCS tend to occur at a younger age, recur more and act more aggressively (4-7). These OKCs occur after age five but peak in the second and third decade. Occasionally, they do not occur until after age 40. Complete surgical removal is the treatment of choice for OKCs regardless of whether or not the patient has the syndrome. Surgery includes enucleation, curettage, enucleation and peripheral ostectomy, or resection depending on the radiographic presentation, location and clinical behavior (4-7). Surgery combined with Carnoy’s solution or liquid nitrogen has been effective in reducing the recurrence rate (7). At times, adjacent or associated teeth are extracted in the interest of complete removal. Some investigators advocate marsupialization and occasionally resection of the more aggressive cysts that tend to perforate buccal and lingual bone (7). OKCs in general, and those associated with NBCCS in particular, are known to have a high recurrence rate, ranging from 13% to 60% (4-7).
Approximately 65 to 80% of the patients with NBCCS present with palmar and/or plantar pits which are around one to two millimeters in size (4, 6); there may be three or more pits. The face is characterized by hypertelorism, frontal bulging, sunken eyes, and heavy and fused eyebrows. Anomalies of the vertebrae and ribs and many other bone abnormalities may be associated with the syndrome. Calcification of the falx cerebri is pathognomonic. There is frontal and temporoparietal bossing, a broad nasal root, prominent supra-orbital ridges and a degree of mandibular prognathism. Short fourth metacarpals are common.
References
- Langlais RP, Langland OE, Nortje CJ. Diagnostic imaging of the jaws. Williams & Wilkins. 1995. Pages 367-370.
- Ozkan Y, Varol A et al. Clinical and radiological evaluation of cherubism: a sporadic case report and review of the literature. Int J Pediatr Otorhinolaryngol. 2003; 67:1005-1012.
- Yamazaki H, Ota Y et al. Brown tumor of the maxilla and mandible: progressive mandibular brown tumor after removal of parathyroid adenoma. J Oral Maxillofac Surg. 2003; 61:719-722.
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004; 6(6):530-9
- Goldberg LH, Bahar F, Weiss G, Blaydorn L, Jameson G, Von Hoff D. Basal cell nevus syndrome: a brave new world. Arch Dermatol. 2010;146(1):17-19
- Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997; 69(3):299-308.
- Oda D, Rivera V et al. Odontogenic keratocyst: the northwestern USA experience. J Contemp Dent Pract. 2000 Feb 15; 1(2): 60-74.